Pompejeva bolezen: simptomi, diagnoza, zdravljenje
Trenutno so znanstveniki identificirali in preučevali na stotine dednih patologij, ki jih praktično ni mogoče zdraviti in povzročajo najnevarnejše posledice. To vključuje redko Pompejevo bolezen, ki jo zdravimo z encimsko nadomestno terapijo. In kljub dejstvu, da je bolezen redka, poskusite razumeti, kaj so vzroki in kakšni so simptomi te nevarne patologije. 
Ogled zgodovine medicine
Za razliko od drugih genetskih bolezni je Pompejevo bolezen prvič odkril pred manj kot stoletjem (1932) nizozemski znanstvenik, po katerem je bil imenovan. I. Pompe je opisal v svojih znanstvenih spisih to bolezen kot dedno bolezen, za katero je značilna kršitev presnove glikogena v telesu. Zaradi takšnih patoloških sprememb so mišična in živčna tkiva motena.
Ad
Ker je identifikacija bolezni v medicini zabeležila nekaj več kot 50 primerov. Hkrati je število žensk in moških skoraj enako. Izid bolezni je v vsakem primeru drugačen in je odvisen od stopnje in oblike patološkega procesa.
Glavni vzroki bolezni
Glavni sprožilec te bolezni je mutacija gena GAA, ki se nahaja na 17. človeškem kromosomu. Vsebuje sekvenčno kodo 4 - glikozide, kislinsko alfa-1 ali maltazo. Izdelava teh ključnih encimov je neposredno odvisna od postopka razdelitve glikogena v njegove sestavne dele, ki se zaradi naslednjih procesov razgradijo v glukozo. Posledica je odsotnost enostavnih segmentov molekul glikogena izmenjavo energije v celicah organizma.
Ad
Prisotnost več oblik bolezni je posledica stopnje mutacije dednega gena. Ko je nosilna koda delno spremenjena, se lahko proces cepitve glikogena upočasni, kar pojasnjuje pojavnost Pompejeve bolezni pri odraslih.
Ko je gen popolnoma mutiran, se prvi simptomi patologije manifestirajo v zgodnjem otroštvu, pogosteje od prvih dni življenja. Zgodnja diagnoza bolezni in pravilna identifikacija njene oblike sta pomembni za izbiro metode zdravljenja in prognoze za bolnika.
Značilnosti bolezni
Izraz pompejeve bolezni pri otrocih in odraslih temelji na klasični glikogenozi, pri kateri skeletne mišice, miokard in živčni sistem trpijo zaradi presežka glikogena. Slednji se ne razgrajuje v telesu in se nabira v celicah mišičnega tkiva, jeter in miokarda. Zato patologija v prisotnosti bolezni uničuje predvsem te organe. V odsotnosti pravočasnega zdravljenja se pri bolnikih pojavijo druge komorbidne bolezni, ki so posledica presnovnih motenj.
Prav tako je treba omeniti, da se genetski prenos mutiranega gena v Pompejevi bolezni izvaja avtosomno recesivno in prekomerno kopičenje glikogena v lizosomih povzroči nastanek velikih vakuol. Posledično so celice s tako velikimi formacijami poškodovane in te spremembe so že nepovratne. 
Razvrstitev glikogenoze 2. vrste
Bolezen, imenovana Pompejeva bolezen, ni nič drugega kot podedovana glikogenoza tipa 2. t Patologija v medicinski praksi je razdeljena na naslednje oblike:
Ad
- Zgodnji infantilni. Po zakonu se ta oblika šteje za najtežjo in nevarnejšo. To se kaže v otrocih v prvih mesecih življenja in hitro napreduje. Pri diagnosticiranju pri dojenčkih se odkrijejo bolezni srca, poškodbe jeter in znaki miopatije. Zaradi hitrega razvoja bolezni je prognoza za bolnike precej negativna. V večini primerov je prišlo do smrti otrok do enega leta zaradi respiratornega ali srčnega popuščanja.
- Pozni infantilni. Za to obliko je značilen razvoj simptomov pri otrocih, starih od 12 do 36 mesecev. Stopnja razvoja bolezni Pompe v takih primerih je nekoliko nižja kot pri zgodnji otroški obliki. V tem primeru je glavna lezija srčna mišica. Otroci umrejo srčno popuščanje v adolescenci.
- Mladoletnik. Simptomi bolezni se kažejo v starosti 6-10 let v obliki poškodbe miokarda. Potek bolezni je počasen. Napoved - smrtnost v starosti 18-20 let.
- Odrasli. V tej obliki Pompejeva bolezen kaže simptome pri bolnikih, ki spadajo v starostno skupino 20-40 let. Glavni simptom je počasi razvijajoča se miopatija. V tem primeru so motnje jeter in miokarda redko fiksirane. Pogosteje gre za manjše spremembe pri delu teh organov. Prognoza za to obliko bolezni je najbolj ugodna. Večina bolnikov živi v starosti in le v posameznih primerih je možna zgodnja smrt zaradi bolezni srca ali respiratorna odpoved.
Sodobni znanstveniki pojasnjujejo variabilnost oblik bolezni s stopnjo mutacije ustreznega gena. Ni jasnejše slike in razlogov za razvoj ene ali druge oblike bolezni, saj je ta sindrom zelo redka.
Ad
Klinična slika patologije
Glavne znake manifestacije bolezni je težko konkretizirati, saj so popolnoma odvisne od oblike patološke motnje. Zato je pravilneje upoštevati klinične manifestacije glede na to merilo. 
Simptomi zgodnje infantilne oblike
V zgodnji infantilni obliki patologijo odkrijejo lokalni pediatri, ki skrbijo za dojenčka. Pri pregledu majhnega bolnika v prvih mesecih beležimo povečanje jeter in zamudo pri psihomotoričnem razvoju. Nekoliko pozneje se simptomi dopolnijo s težavami pri zaužitju hrane zaradi izrazite slabosti sesalnih mišic. Razkrila je tudi disfagijo in po minimalnem času popolno hipotrofijo. Ker je bolezen redka, pediatri niso vedno sposobni pravočasno postaviti pravilne diagnoze. Navsezadnje so simptomi in fotografije Pompejeve bolezni pri otrocih podobni mnogim drugim boleznim. Kasneje, drobtine prihajajo z dihanjem in srčnim popuščanjem in posledično s smrtjo.
Manifestacije pozne infantilne in mladostne oblike
V pozni in mladostni patologiji je klinična slika zelo podobna. Edina razlika je v času nastanka prvih znakov, in sicer odkritja šibkosti mišic. Pozneje se simptomi dopolnjujejo z motnjami srčne mišice. Nekaj mesecev po pojavu prvih znakov zdravniki diagnosticirajo nastanek mišične distrofije skeletnih mišic in kardiomegalije. Istočasno se v ozadju znakov, ki so se pojavili, ki se lahko odkrijejo z ultrazvokom in palpacijo, občutno poveča vranica in jetra.
Ad
Trajanje bolezni v vsakem primeru je lahko različno, povprečno obdobje pa je 9-12 let. Istočasno se lahko pojavijo posredni simptomi patologije v obliki glavobolov, nočno apnejo in pogostih bolezni dihal različne kompleksnosti. Pomanjkanje terapije je smrtno posledica srčnega popuščanja.
Simptomi odrasle oblike
V odrasli obliki je desetkrat težje prepoznati bolezen, ker se klinični znaki pojavijo počasi in se lahko razmazajo s simptomi kroničnih bolezni. Pri tej obliki pompejeve bolezni je mišična šibkost pogosto pripisana utrujenosti ali preobremenitvi. Po nekaj letih se skolioza ali lardoza, ki jo povzroči šibkost mišične hrbtenice, pridruži primarnim simptomom. V redkih primerih s popolnim pregledom bolnikov razkrivamo blage znake srčnega popuščanja.

Patologija se lahko razvija več desetletij brez ogrožanja kakovosti življenja. Zato tudi v odsotnosti terapije večina bolnikov živi v starosti.
Diagnostične metode
Kljub temu, da je Pompejeva bolezen precej redka, so jo znanstveniki podrobno preučili. Vendar pa ni posebne metode, ki bi podala podroben odgovor o bolnikovem stanju. Zato je v medicinski praksi diagnoza "glikogenoza tipa 2" narejena na podlagi celovite študije, ki vsebuje rezultate:
- biokemična analiza krvi;
- pregled in zbrana zgodovina;
- študije biopsije mišic;
- EKG;
- EchoCG in druge laboratorijske ter instrumentalne metode.
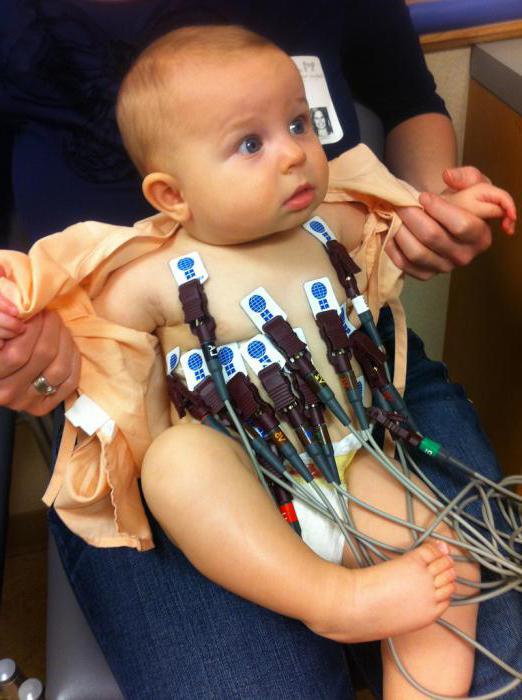
Diagnoza Pompejeve bolezni se začne že pri bolniku. Zdravnik razkrije slabost mišične mišice, kar potrjujejo rezultati zbrane zgodovine. Pri takih simptomih se bolniku priporoča izdelava kardiograma, ki jasno kaže zmanjšanje intervala PQ, kot tudi širitev kompleksa QRS, ki ga povzroča povečanje miokarda. Motnje pri delu srčna mišica To je tudi vzrok inverzije T-valov, ki se kaže kot posledica povečanja trajanja ventrikularne repolarizacije. Treba je omeniti, da takšna študija lahko potrdi prisotnost Pompejeve bolezni le pri otrocih, saj v odrasli obliki morda ne pride do kršitve miokarda.
Biokemijska laboratorijska preiskava pacientove krvi omogoča potrditev ali zavrnitev predhodne diagnoze specifičnih in posrednih znakov. V specifičnem krvnem testu določimo aktivnost 4-glikozidov in alfa-1 v plazmi. Z zmanjšanjem normalnih kazalnikov takšna analiza potrjuje prisotnost bolezni. Kot pri posrednih krvnih preiskavah je pri Pompejevi bolezni zabeleženo močno povečanje kreatin fosfokinaze.
Ko je histologija mišične biopsije v laboratoriju pokazala v miocitih več vključkov glikogena. V Pompejevi bolezni je slika takih patologij pod mikroskopom podobna penastim celicam.
Končna točka pri postavitvi takšne diagnoze lahko postavi zdravnika genetičnega zdravnika po študiji, ki jo je izvedel z zaporedjem prizadetega gena.
Terapija in prognoza bolezni
Danes je edina možnost zdravljenja Pompejeve bolezni encimska nadomestna terapija. To vam omogoča, da zapolnite primanjkljaj 4-glikozidi in alfa-1. Potrebne priprave so danes narejene le v ZDA, njihova cena pa je precej visoka. 
Potek terapije, ki traja 12 mesecev, bo stala približno 100-400 tisoč dolarjev, to pa ni na voljo vsem. Toda tudi če je možno kupiti potrebna zdravila, je nemogoče napovedati izid zdravljenja, ker je individualen.
Brez ustrezne terapije je prognoza za bolnike z infantilno in mladostno obliko bolezni neugodna. Pri patologiji odraslega je prognoza dvoumna.